

Summary:
- XenpozymeTM (olipudase alfa-rpcp) approved by FDA as first disease-specific treatment for ASMD (non-CNS manifestations)
- Xenpozyme is the first approved medication to treat symptoms that are not related to the central nervous system in patients with ASMD.
- Olipudase alfa is indicated as an enzyme replacement therapy for the treatment of non-Central Nervous System manifestations of Acid Sphingomyelinase Deficiency (ASMD) in pediatric and adult patients with type A/B or type B.
The U.S. Food and Drug Administration (FDA) has approved XenpozymeTM (olipudase alfa-rpcp) for the treatment of non-central nervous system (non-CNS) manifestations of acid sphingomyelinase deficiency (ASMD) in adult and pediatric patients. Xenpozyme is the first therapy indicated specifically for the treatment of ASMD, and is currently the only approved treatment for this disease.
About XenpozymeTM
- API– Olipudase alfa-rpcp
- Description: Olipudase alfa is recombinant human acid sphingomyelinase. Olipudase alfa was first approved in Japan on March 28, 2022 and by the European Commission on June 28, 2022. It is the first and only enzyme replacement therapy for the treatment of Acid Sphingomyelinase Deficiency (ASMD), also known as Niemann–Pick disease, in both regions. ASMD is a rare lysosomal storage disease caused by mutations in the SMPD1 gene, leading to a deficiency in acid sphingomyelinase and the abnormal accumulation of the primary ASM substrate, sphingomyelin. Olipudase alfa works to hydrolyze sphingomyelin accumulated in body tissues, such as the lungs, liver, spleen, kidneys, and bone marrow.
- Indication-Olipudase alfa is indicated as an enzyme replacement therapy for the treatment of non-Central Nervous System manifestations of Acid Sphingomyelinase Deficiency (ASMD) in pediatric and adult patients with type A/B or type B.
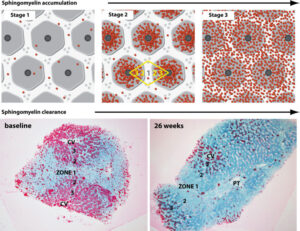
Clearance of Hepatic Sphingomyelin by Olipudase Alfa Is Associated With Improvement in Lipid Profiles in Acid Sphingomyelinase Deficiency.
- Biologic Classification :Protein Based Therapies
- Mechanism of action: ASMD is an autosomal recessive genetic disorder caused by different mutations in the SMPD1 gene that encodes acid sphingomyelinase. Historically, ASMD has been called Niemann-Pick disease (NPD), with different classifications based on disease onset and severity. NPD type A (NPD-A) refers to the severe early-onset form, infantile neurovisceral ASMD and NPD type B (NPD-B) is referred to as the later-onset, chronic visceral form of ASMD. Chronic neurovisceral ASMD (NPD-A/B) is a phenotype with intermediate severity. ASMD has a broad spectrum of disease severity and neurological and non-neurological manifestations; thus, it is difficult to classify different types of ASMD using the former classification system of ASMD (A, B, A/B). Acid sphingomyelinase typically breaks down metabolically-related lipids such as sphingomyelin in various cell types, such as the monocytes, macrophages, and hepatocytes. The deficiency of acid sphingomyelinase thus leads to the accumulation of these lipids in body tissues, causing progressive cell and tissue damage and impairing organ functioning. Olipudase alfa is recombinant human acid sphingomyelinase that hydrolyzes sphingomyelin (SM), preventing its accumulation in body organs. As an enzyme replacement therapy, it is an exogenous source of acid sphingomyelinase
- Metabolism: Olipudase alfa is a recombinant human enzyme and is expected to be eliminated via proteolytic degradation into small peptides and amino acids.
- Half-life: After administration of 3 mg/kg olipudase alfa once every two weeks in adults with ASMD, the mean terminal half-life (t1/2) ranged from 31.9 to 37.6 hours
Bill Sibold
Executive Vice President, Head, Specialty Care at Sanofi
“Sanofi teams have been dedicated to bringing hope to patients living with ASMD and their families. This is a devastating and extremely rare disease that affects both children and adults. The approval of Xenpozyme represents the culmination of bold work done in research and development, and our unwavering commitment to this historically overlooked community.”
ASMD, historically known as Niemann-Pick disease types A, A/B, and B, is an extremely rare, progressive genetic disease with significant morbidity and mortality. It has been estimated that there are fewer than 120 patients diagnosed with ASMD in the U.S. Approximately two-thirds of patients with ASMD in the U.S. are pediatric. Signs and symptoms of ASMD can present in infancy, childhood, or adulthood, and may include enlarged spleen or liver, difficulty breathing, lung infections, and unusual bruising or bleeding, among other disease manifestations. Until now, management of ASMD included supportive care to address the impact of individual symptoms and careful monitoring to detect potential disease complications.
David Guy
Parent to Kaila, age 16, living with ASMD
“As young parents, it was initially devastating to me and my wife when our daughter, Kaila, received her diagnosis of ASMD. We faced so many unknowns when we first heard the diagnosis: what does this mean, how will this affect her, and most importantly what hope is there for a treatment option? We were grateful to find hope when we enrolled Kaila in the clinical trials for olipudase alfa.”
In the U.S., Xenpozyme received Breakthrough Therapy designation, which expedites the development and review of drugs intended to treat serious or life-threatening diseases and conditions. The FDA evaluated Xenpozyme under Priority Review, which is reserved for medicines that represent potentially significant improvements in efficacy or safety in treating serious conditions.. In March 2022, Xenpozyme was approved in Japan under the SAKIGAKE (or “pioneer”) designation, marking the first approval for olipudase alfa anywhere in the world. In June 2022, the European Commission (EC) approved Xenpozyme for use in Europe.
ASMD represents a spectrum of disease, with two types that may represent opposite ends of a continuum referred to as ASMD type A and ASMD type B. ASMD type A/B is an intermediate form that includes varying degrees of central nervous system (CNS) involvement.
ASCEND and ASCEND-Peds clinical trials showed that Xenpozyme improved lung function and reduced spleen and liver volumes in adults and children
The approval is based on positive data from the ASCEND and ASCEND-Peds clinical trials, in which Xenpozyme showed clinically relevant improvement in lung function (as measured by diffusing capacity of the lung for carbon monoxide, or DLco) and platelet count, and reduction of spleen and liver volumes, with a demonstrated safety profile.
Melissa Wasserstein
MD, Pediatric Genetic Medicine, Albert Einstein College of Medicine and the Children’s Hospital at Montefiore
“ASMD is an extremely rare, progressive, and potentially fatal genetic disease that impacts children and adults around the world. Until now, those living with ASMD have had no FDA-approved treatment to combat this devastating condition. I’m proud of the work that has been done and look forward to witnessing the impact that this treatment may have on those living with ASMD.”
The ASCEND trial evaluated the efficacy and safety of Xenpozyme; 31 adult patients with ASMD type A/B or type B were randomized to receive Xenpozyme or placebo for 52 weeks (primary analysis). In the trial, Xenpozyme improved lung function, assessed as the percent change from baseline to Week 52 in predicted diffusing capacity of the lung for carbon monoxide (DLco), and reduced spleen volume, evaluated as percent change from baseline in multiples of normal (MN).
- Twelve (12) patients treated with Xenpozyme had a mean change in percent predicted DLco from baseline (49.1%) to Week 52 (59.4%). This change represents a 23.9% relative improvement compared to a 3% improvement in DLco from baseline in the 17 patients from the placebo group (48.5%) to Week 52 (49.9%). The difference between the two arms (20.9%) was nominally statistically significant (p=0.0003).
- Thirteen (13) patients treated with Xenpozyme had a mean reduction in spleen volume by 38.9% from baseline (11.5 MN) to Week 52 (7.2 MN) compared to a mean increase by 0.5% for the 17 patients in the placebo group from baseline (11.2 MN) to Week 52 (11.2 MN). The difference between the two arms (39.4%) was nominally statistically significant (p<0.0001).
- Twelve (12) patients treated with Xenpozyme had a mean reduction in liver volume by 26.5% from baseline (1.4 MN) to Week 52 (1.0 MN) compared to a mean decrease of 1.8% for the 17 patients in the placebo group from baseline (1.6 MN) to Week 52 (1.6 MN). The difference between the two arms (24.7%) was nominally statistically significant (p<0.0001).
- Thirteen (13) patients treated with Xenpozyme had a mean improvement in platelet count by 18.3% from baseline (109.3×109/L) to Week 52 (126.4×109/L) compared to increase by 2.7% for the 16 patients in the placebo group from baseline (115.6×109/L) to Week 52 (120.2×109/L). The difference between the two arms (15.6%) was nominally statistically significant (p=0.0280).
- All ASCEND patients treated with Xenpozyme showed improvement in key endpoints (DLco and spleen and liver volume).
- Most frequently reported adverse drug reactions in adults (incidence ≥10%) were headache, cough, diarrhea, hypotension, and ocular hyperemia.
The single-arm ASCEND-Peds trial studied 8 pediatric patients younger than 12 years of age with ASMD type A/B or type B who all received Xenpozyme, with a primary objective of evaluating the safety and tolerability of Xenpozyme for 64 weeks. All patients completed the study and continued in an extension trial. The ASCEND-Peds trial also explored efficacy endpoints of progressive lung disease, spleen and liver enlargement, and platelet count. After one year of treatment (52 weeks):
- Three (3) patients who were able to perform the test at baseline treated with Xenpozyme had a mean relative improvement of 45.9% in percent predicted DLco from baseline (48.5%) to Week 52 (70.9%) (children over the age of five were assessed if they were able to perform the test).
- Eight (8) patients treated with Xenpozyme had mean reduction in spleen volume by 46.7% from baseline (18.3 MN) to Week 52 (9.5 MN).
- Eight (8) patients treated with Xenpozyme had a mean reduction in liver volume by 38.1% from baseline (2.5 MN) to Week 52 (1.6 MN).
- Seven (7) patients treated with Xenpozyme had a mean improvement in platelet count by 37.6% from baseline (136.7×109/L; n=8) to Week 52 (184.5×109/L).
- Serious adverse reactions of anaphylactic reaction were reported in 2 (25%) Xenpozyme-treated pediatric patients.
- Treatment-related serious adverse reactions, hypersensitivity reactions including anaphylaxis, and infusion associated reactions occurred within 24 hours of infusion and were observed in a higher percentage of pediatric patients than in adult patients.
- Most frequently reported adverse drug reactions in pediatric patients (incidence ≥20%) were pyrexia, cough, diarrhea, rhinitis, abdominal pain, vomiting, headache, urticaria, nausea, rash, arthralgia, pruritus, fatigue, and pharyngitis.
About Acid Sphingomyelinase Deficiency (ASMD)
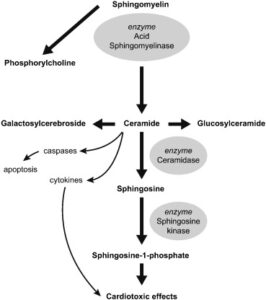
Acid Sphingomyelinase Deficiency (ASMD)
Acid sphingomyelinase deficiency (ASMD) is a rare progressive genetic disorder that results from a deficiency of the enzyme acid sphingomyelinase, which is required to break down (metabolize) a fatty substance (lipid) called sphingomyelin.
- Protruding abdomen. due to enlarged liver. and/or spleen.
- Coughing and. difficulty breathing.
- Easy bruising. and bleeding.
- Abdominal pain. and diarrhea.
A scientific innovation for patients living with ASMD
Xenpozyme, a hydrolytic lysosomal sphingomyelin-specific enzyme replacement therapy, is designed to replace deficient or defective acid sphingomyelinase (ASM), an enzyme that allows for the breakdown of the lipid sphingomyelin. In individuals with ASMD, the deficiency in the ASM enzyme leads to sphingomyelin accumulation in various tissues. Xenpozyme is not expected to cross the blood-brain barrier or modulate CNS manifestations of ASMD. Xenpozyme has not been studied in patients with ASMD type A.
Xenpozyme is adminstered intravenously every two weeks, and its administration requires a dose escalation phase followed by a maintenance phase.
Xenpozyme is expected to be available in the U.S. in the coming weeks. The U.S. list price, or wholesale acquisition cost, of Xenpozyme is $7,142.00 per vial. Actual patient out-of-pocket costs may be lower, as the list price does not reflect insurance coverage, co-pay support for eligible patients, or financial assistance from patient support programs.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people’s lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Weblink: https://www.chemrobotics.com
- AgroPat Lite– Access 5500 pesticides with chemistry, Biology, Regulatory, and IP info. Covers the product information including formulation, combination, developer, innovator, existing intellectual property, regulatory requirement, biology data including spectrum, MOA, DFU, toxicity profile, and safety. (Designed for Business Development function)
-
-
- AgroPat Ultimate– In detailed Access 5500 pesticides with chemistry, Biology, Regulatory, and IP info. (Designed for Research & Development function)
- Indian Medicine Database –Approved Drugs, Medical Devices, Approved Regenerative Medical Products
- Weblink: https://imd.chemrobotics.com/
- Indian Pesticide Database (IPD)– All Indian Approvals, e.g. 9(3) and 9(4), etc.
- Global Agro Product Directory(More than 55countries approved product info. with relevant documents such as label, factsheet and monograph)
- Weblink: https://www.chemrobotics.com/pesticides-directory/
- Global MRL Database(More than 85 countries MRL info.)
- Jarvis– A Competitor Patents Watch Database for Agrochemical
- Technical Routes(More than 15000 routes of synthesis for Agrochemical & Pharmaceutical)
- Technical Suppliers(Provides technical supplier information)
- Company Directory– KSM Supplier(s) Database — More than 10 K Companies listed from Pharma / Agrochemical / Fine Chemical Domain with their product offering in Pharma / Agrochemical / Fine Chemical segment,
- Weblink: https://companydirectory.chemrobotics.com
- ChemRobotics SPC Database– Provides Patent SPC data Europe
- PharmVetPat –Access chemistry including ROS, KSM, Intermediate, Biology, Regulatory, and IP info for all pharm molecules.
-
- Weblink: https://chemroboticspharma.com/pharmVetPat