

Synopsis :
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
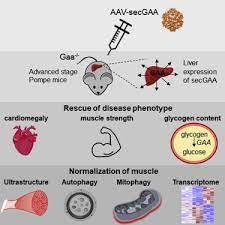
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen – the stored form of glucose – breaks down to release glucose into the bloodstream to be used as fuel for the cells.

Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
About Pompe disease
Pompe disease is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body’s cells. The accumulation of glycogen in certain organs and tissues, especially muscles, impairs their ability to function normally.
Researchers have described three types of Pompe disease, which differ in severity and the age at which they appear. These types are known as classic infantile-onset, non-classic infantile-onset, and late-onset.
The classic form of infantile-onset Pompe disease begins within a few months of birth. Infants with this disorder typically experience muscle weakness (myopathy), poor muscle tone (hypotonia), an enlarged liver (hepatomegaly), and heart defects. Affected infants may also fail to gain weight and grow at the expected rate (failure to thrive) and have breathing problems. If untreated, this form of Pompe disease leads to death from heart failure in the first year of life.
The non-classic form of infantile-onset Pompe disease usually appears by age 1. It is characterized by delayed motor skills (such as rolling over and sitting) and progressive muscle weakness. The heart may be abnormally large (cardiomegaly), but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems, and most children with non-classic infantile-onset Pompe disease live only into early childhood.
The late-onset type of Pompe disease may not become apparent until later in childhood, adolescence, or adulthood. Late-onset Pompe disease is usually milder than the infantile-onset forms of this disorder and is less likely to involve the heart. Most individuals with late-onset Pompe disease experience progressive muscle weakness, especially in the legs and the trunk, including the muscles that control breathing. As the disorder progresses, breathing problems can lead to respiratory failure.
What are the types of Pompe disease?
There are three types of Pompe disease:
- Classic infantile-onset appears within a few months of birth.
- Non-classic infantile-onset appears at about 1 year of age.
- Late-onset appears later in a child’s life, or even into the teen years or adulthood.
Who gets Pompe disease?
Since this is a genetic condition, the people who get this disease inherit it from a parent. It is common, however, that neither parent shows any symptoms. The disease is rare. In the United States, only 1 person in 40,000 is affected by Pompe disease. It can affect both males and females of all ethnic groups.
What are the symptoms of each type of Pompe disease?
Symptoms can be a bit different, depending on when the disease makes itself present. In infants, symptoms include the following:
Classic type:
- Weak muscles
- Poor muscle tone
- Enlarged liver
- Failure to gain weight and grow at the expected rate (failure to thrive)
- Trouble breathing
- Feeding problems
- Infections in the respiratory system
- Problems with hearing
Non-classic type:
- Motor skills delayed (such as rolling over and sitting)
- Muscles get steadily weaker
- Abnormally large heart
- Breathing problems
Late-onset type, which includes adult-onset:
- The legs and the trunk get steadily weaker.
- Breathing problems
- Enlarged heart
- Increasing difficulty in walking
- Muscle pain over a large area
- Loss of the ability to exercise
- Falling often
- Frequent lung infections
- Shortness of breath when the person pushes himself or herself
- Headaches in the morning
- Becoming tired during the day
- Losing weight
- Cannot swallow as easily as before
- Irregular heart beat
- Increased difficulty hearing
- Higher levels of creatine kinase (CK) , an enzyme that helps the body’s functions work and provides energy to cells
How is Pompe disease diagnosed?
A blood sample is taken and enzymes in the blood are studied and counted. Also, there are tests such as sleep studies, breathing tests to measure lung capacity, and electromyography (a test that measures how well the muscles work).
A blood sample is taken and enzymes in the blood are studied and counted. Confirmation is made via DNA testing. Other tests include:
- Taking of a complete patient and family history
- Breathing tests to measure lung capacity (pulmonary function tests)
- Electromyography (a test that measures how well the muscles work) and MRIs
- Heart studies, including X-rays, electrocardiogram and echocardiogram
- Sleep studies
A prenatal diagnosis may be done for pregnant women at risk.
How is Pompe disease treated?
Enzyme replacement therapy (ERT) is an approved treatment for all Pompe patients. A drug called alglucosidase alfa is given intravenously (through the patient’s vein). It is a genetically engineered enzyme that acts like the naturally occurring acid alfa glucosidase enzyme.
Specialist teams (heart doctors, respiratory therapists, neurologists, etc.) can treat symptoms and offer supportive care for those with Pompe disease. Ask your doctors about details for each specific case.
What are the complications of Pompe disease?
Without treatment, infants with Pompe disease will die. Many of the people with Pompe disease have respiratory (breathing) problems, heart problems, and almost all are plagued with muscle weakness. Most people will have to use oxygen and wheelchairs at some point.
How can Pompe disease be prevented?
As this is a genetic disease it cannot currently be prevented. Supportive treatment and care are available.
What is the outlook for people with Pompe disease?
Patients with either type of infantile-onset Pompe disease may have their lives prolonged with early detection and treatment. However, both of these types of Pompe disease often are fatal. Patients with classic infantile-onset type rarely live past 1 year of age. Patients with non-classic infantile–onset type may live to early childhood. Children with late-onset types of Pompe disease can live longer as the disease progresses more slowly.